Professorship of Bioinformatics
Prof. Dr. Heinrich Sticht
Kontakt
Research Focus
Protein-protein interactions play a crucial role for the transduction of information in biological systems. The identification of the underlying principles of molecular recognition is important for the understanding of regulatory mechanisms as well as for the prediction of novel, physiologically relevant protein interactions. The bioinformatics group is primarily interested in investigating molecular interactions by a variety of computational tools (e.g. sequence data analysis, molecular modeling, and molecular dynamics).
Molecular dynamics simulations are used to study the dynamics of viral proteins, the conformational transitions of human proteins (e.g. Alzheimer Aβ-Amyloid), or the effect of covalent modifications on molecular recognition processes. Molecular modeling is used to generate the structure of isolated proteins or biomolecular complexes and provides the basis for a molecular understanding of mutational effects on protein stability and binding properties. In addition, sequence based methods are developed that allow an improved detection of functional linear interaction motifs. Such motifs play an important role for the interactions of numerous pathogens with the target molecules of their host.
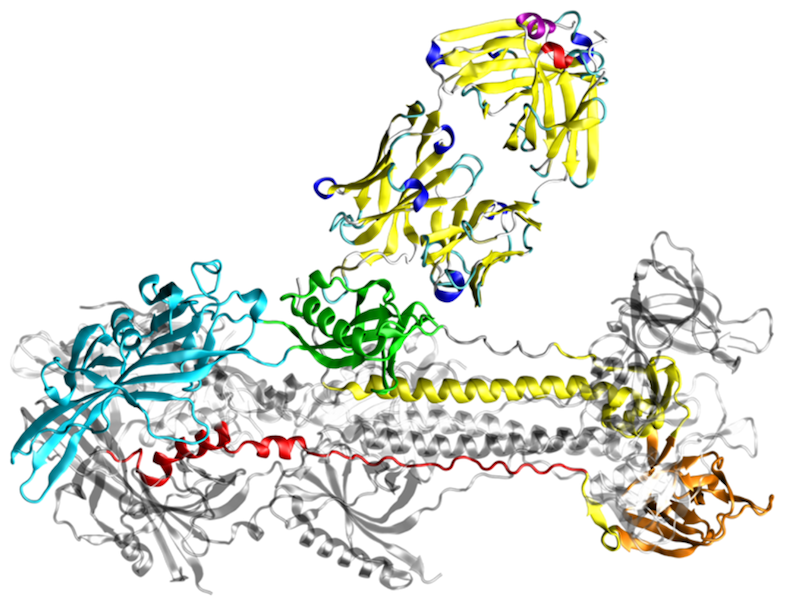
Specific interactions with host proteins are pivotal for a successful infection by a pathogen. Our research focuses on the prediction and structural characterization of host-pathogen protein interactions using computational tools.
In most research projects, we use a combination of molecular modeling, docking, and molecular dynamics simulations. The latter technique provides information about the conformational stability and energetics of an interaction that can hardly be deduced from static structures alone. We use such simulations, for example, to identify and optimize peptides that bind to viral fusion proteins from HIV-1 or SARS CoV-2 and thus block viral infection. Another focus is the investigation of the interaction of herpesviral proteins with their cellular targets.
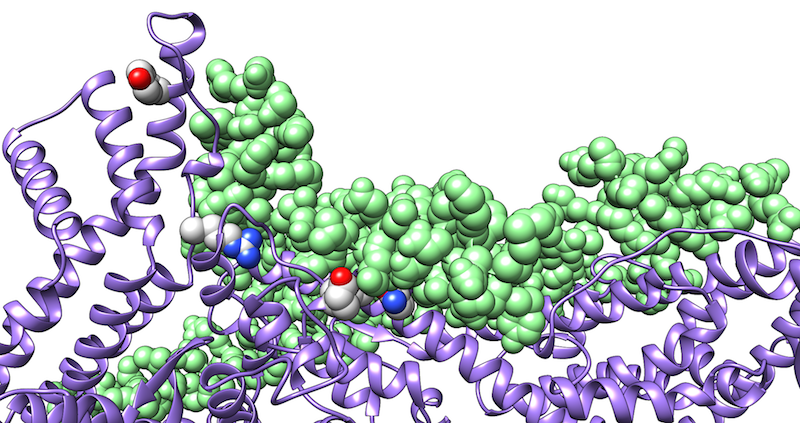
Protein conformational diseases are unique since they result from a drastic change in protein three-dimensional structure. Most often, the change in conformation involves a structural conversion from primarily α-helical conformation with good solubility to an insoluble β-sheet conformation. Cells have evolved mechanisms to clear these insoluble deposits; however, once clearance pathways are overloaded, these proteins are deposited in the form of insoluble intracellular inclusions or extracellular plaques. Protein deposits or aggregates are also hallmark of many neurodegenerative diseases.
The most prevalent neurodegenerative disease is Alzheimer’s disease, which is characterized by extracellular protein deposition of the peptide fragment Aβ from the amyloid precursor protein, and intracellular tau-containing filaments, called neurofibrillary tangles. The 3D structure of the Aβ deposits revealed the overall topology of the fibrils, but gives only limited information about the role of individual residues for fibril formation. The latter type of information, however, is important for the development of novel drugs that are capable of preventing aggregation or of solubilizing aggregates by targeting those residues that represent the hot spots of binding affinity in the fibrillar structure. We address this point by molecular dynamics simulations of Aβ oligomers and thermodynamic analyses of the aggregation interfaces. In addition, we investigate the effect of different solvent environments on the conformational stability of such Aβ oligomers.
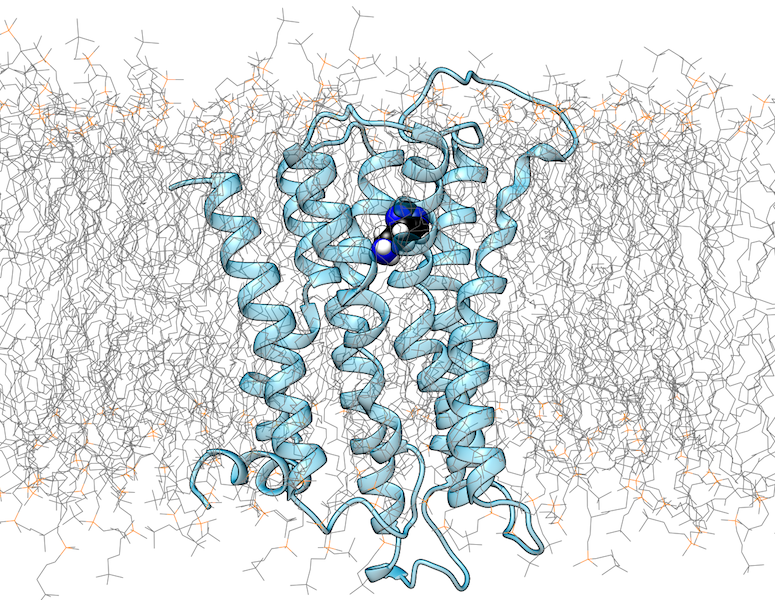
G-protein coupled receptors (GPCRs) are transmembrane proteins that recognize extracellular ligands and thereby trigger intracellular signaling processes. There are now more than 100 GPCR crystal structures that provide valuable insights into the structural properties of this protein family. However, the current understanding of the structure and function of GPCRs is far from complete.
A major reason for this is that GPCRs occur in active and inactive conformations, of which usually only one state could be crystallized. In addition, the structure is influenced by the interaction with intracellular binding partners (IBPs) such as G-proteins or β-arrestin. The extracellular ligand binding pocket also exhibits a certain structural plasticity, which is particularly relevant for drug development. Since the crystallization of GPCRs in different activation states or in complexes with different ligands is experimentally very challenging, computer-assisted methods are of outmost importance for the investigation of these aspects.
We use methods of molecular modelling and molecular dynamics to study the structure of GPCRs in complex with different small molecule ligands or intracellular interaction partners. Aspects investigated include the prediction of the binding modes of small molecule ligands, conformational changes in GPCRs as a result of ligand binding, and the influence of mutations on GPCR function and interaction. In addition to conventional MD simulation methods, computationally demanding metadynamics simulations are also used.
In addition to GPCRs, we also investigate other classes of membrane receptors using similar methodological approaches. Systems studied include the glycine receptor, at which we characterize the binding site of saccharides as allosteric modulators. In the case of the macrophage surface receptor Mincle, we are investigating the binding of synthetic glycolipids, which should support the long-term development of better adjuvants for vaccines.
Changes in pH regulate many biological processes in bacteria, viruses, vertebrates, and plants. For example, some bacteria are able to survive the acidic conditions in the stomach of their host by using acid-activated chaperones, which protect substrate proteins from aggregation. In viruses, some of the fusion proteins that mediate cell entry were described to act pH-dependently. Other proteins in vertebrates undergo pH changes on their way through the endoplasmic reticulum and the Golgi apparatus. In order to mimic pH-titration experiments, we investigate pH-dependent proteins by conducting molecular dynamics simulations, in which pH is gradually changed. This method allows the calculation of titration curves and pKa values of ionizable groups. By using this strategy, we investigate on an atomic level the effects of pH changes which affect protein local conformations, macromolecular assemblies as well as structural stability.
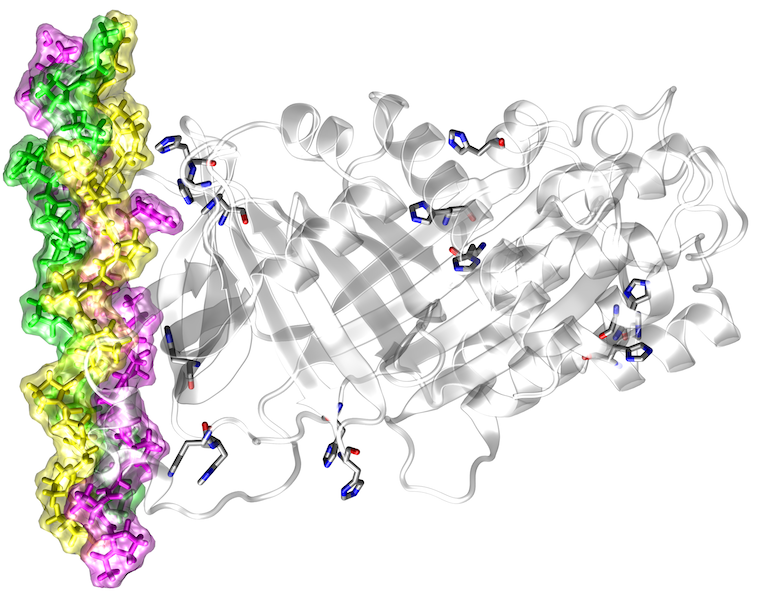
High-throughput DNA sequencing studies revealed a large number of genetic variants between individuals. Many of these sequence variants lead to amino acid exchanges, some of which are linked to disease. Due to their large number (> 10,000 per genome), it is impossible to characterize all sequence variants by experiment, rendering computational prediction tools of utmost importance for the identification of pathogenic variants. Most of the current methods use evolutionary conservation and other sequence-based features to identify damaging variants, but they cannot predict the effects these variants have on protein function. Despite its innate linkage to function, structural information is yet only considered to a very limited extent in the predictions. In addition, the few existing structure-based prediction methods mainly focus on one distinct aspect of protein structure (e.g. protein stability or protein interactions) and do therefore not allow a comprehensive structural and functional annotation. The aim of the present project is to develop a robust computational framework for a comprehensive structure-based analysis and interpretation of high-throughput sequencing data.